Благодаря значительным достижениям микробиологии, биохимии и эпидемиологии в нашей стране значительно изменилась структура заболеваний населения. Ликвидированы или почти исчезли тяжелые инфекционные заболевания (оспа, чума, холера, трахома, полиомиелит, малярия).
Сведены к минимуму кишечные инфекции.
Но, занимают одно из ведущих мест в структуре наследственной патологии человека, хромосомные болезни. Они у новорожденных детей встречаются с частотой примерно 2,4 случая на 1000 родившихся (по данным цитогенетиче ских исследований частота хромосомной патологии составляет 0,6-1,0%).
Самая высокая частота хромосомной патологии (до 70%) зафиксирована в материале ранних спонтанных абортов. Следовательно, большинство хромосомных аномалий у человека несовместимо даже с ранними этапами эмбриогенеза. Такие зародыши элиминируются во время имплантации (7-14-е дни развития), что клинически проявляется как задержка или выпадение менструального цикла. Некоторая часть эмбрионов гибнет вскоре после имплантации (ранние выкидыши).
Сравнительно немногие варианты числовых аномалий хромосом совместимы с постнатальным развитием и ведут к хромосомным заболеваниям (Кулешов Н.П., 1979).[3]
1.
Хромосомные болезни или хромосомные синдромы – это комплексы множественных врожденных пороков развития, вызываемых числовыми или структурными изменениями хромосом, видимыми в световой микроскоп.
Нарушения в строении хромосом, изменения их количества, генные мутации могут возникать на разных этапах развития организма. Если они возникают в гаметах родителей, то аномалия будет наблюдаться во всех клетках организма (полный мутант). [2]
Если они возникают в процессе эмбрионального развития, хромосомный набор в разных клетках тела будет разным. В процессе развития появляется несколько следующих друг за другом поколений клеток с различными хромосомными наборами. При незначительном количестве аномальных клеток болезни в последующем может и не быть.
К ним относят изменение плоидности, например утроенный набор хромосом (З
При многих из указанных хромосомных изменений развивающиеся из зиготы эмбрион и плод нежизнеспособны. Эти изменения хромосом часто выявляются у абортов (абортированные плоды), а также у новорожденных, которые вскоре после рождения погибают.
Особенности физического и психического развития детей с патологиями зрения
... что дети с депривацией зрения оказываются в стрессовых ситуациях чаще, чем их нормально видящие сверстники. В данном реферате рассмотрим особенности физического и психического развития детей с патологиями зрения, а ... из общего числа глазных заболеваний у детей 2.ОСОБЕННОСТИ ПСИХИЧЕСКОГО РАЗВИТИЯ ДЕТЕЙ С НАРУШЕНИЕМ ЗРЕНИЯ Неудачи и трудности, с которыми ребенок сталкивается в обучении, в игре, в ...
Совместимыми с жизнью являются трисомия по отдельным аутосомам и половым хромосомам, моносомия по половой Х-хромосомсе, а также относительно небольшие структурные изменения хромосом. Однако и при этих изменениях отмечается ряд дефектов в организме проявляющихся порою значительными уродствами.
Хромосомные болезни не передаются по наследству. Кариотип родителей этих больных обычно нормальный, а аномальные хромосомные изменения обычно происходят в гаметах, из которых развивается больной организм. Но, так как именно в хромосомах находится наследственная информация, то хромосомные болезни по механизму своего возникновения следует трактовать как наследственные, поскольку в их основе лежит нарушение аппарата наследственности.
-
Факторы повышенного риска рождения детей с хромосомными болезнями
Имеются экспериментальные данные о влиянии на мутационный процесс таких факторов, как: действие ионизирующих излучении, химических веществ, вирусов. Другими причинами нерасхождения хромосом могут быть: сезонность, возраст отца и матери, порядок рождения детей, прием лекарств во время беременности, гормональные нарушения, алкоголизм. Не исключается до определенной степени и генетическое детерминирование нерасхождения хромосом. Но причины образования геномных и хромосомных мутаций на ранних стадиях развития зародыша до сих пор окончательно не раскрыты. К биологическим факторам повышения риска рождения детей с хромосомными аномалиями может быть отнесен возраст матери. Риск рождения больного ребенка особенно резко возрастает после 35 лет. Это характерно для любых хромосомных болезней, но наиболее четко наблюдается для болезни Дауна. В медико-генетическом планировании беременности особое значение уделяется двум факторам — наличию анеуплоидии по аутосомам у ребенка и возрасту матери старше 35 лет. К кариотипическим факторам риска у супружеских пар относятся: анеуплоидия (чаще в мозаичной форме), робертсоновские транслокации (слияние двух телоцентрических хромосом в области деления), кольцевые хромосомы, инверсии. Степень повышения риска зависит от типа хромосомных нарушений. В настоящее время оценка степени риска уступает более точной пренатальной цитогенетической диагностики эмбриона или плода.
Зависимость частоты рождения детей с хромосомными болезнями от возраста матери
1:500
Изменение структуры и функции семьи в связи с рождением ребенка
... очень актуальная. Цель исследования - изучение изменение структуры и функции семьи в связи с рождением ребенка. Задачи: Рассмотреть стадии развития семьи. Выявить сложности в семье после рождения ребенка. Проанализировать преодоления кризиса в семье в связи с рождением ребенка. Стадии развития семьи. Каждая семья осуществляет свой жизненный цикл. Она проходит ...
25
1:1300
1:500
30
1:1000
1:400
35
1:300
1:200
40
1:100
1:70
45
1:30
1:20
49
1:12
1:8
Медико-генетическое консультирование
медико-генетическая консулътация
1) консультация врача-генетика как врачебное заключение;
2) структурное подразделение в каком-либо звене здравоохранения (при больнице, при объединении, поликлинике и др.).
Показаниями для медико-генетического консультирования являются:
1) рождение ребенка с врожденным пороком развития;
2) установленная или подозреваемая наследственная болезнь в семье в широком смысле слова;
3) задержка физического развития или умственная отсталость у ребенка;
4) повторные спонтанные аборты, выкидыши, мертворождения;
5) близкородственные браки;
6) воздействие подозреваемых на тератогенность или известных тератогенов в первые 3 мес. беременности;
7) неблагополучное протекание беременности. В принципе каждая супружеская пара должна пройти медико- генетическое консультирование до планирования деторождения (проспективно) и безусловно после рождения больного ребенка (ретроспективно).
При выявлении моногенной болезни врач должен предоставить больному или его семье соответствующую генетическую информацию и провести медико-генетическое консультирование. Основа всего — постановка правильного диагноза с обязательным использованием доступных биохимических и молекулярно-генетических методов. Достоверность диагноза играет решающую роль для интерпретации характера передачи наследуемых признаков и расчета риска заболевания у родственников.
-
Болезни, обусловленные нарушением числа хромосом
Болезни, обусловленные нарушением числа хромосом
1.Моносомия – это наличие всего одной из пары гомологичных хромосом (
Трисомии и причины их возникновения
... в соматических клетках человека. В работе рассматриваются наследственные синдромы вызванные численными мутациями хромосом – трисомии (трисомия 21 – синдром Дауна, трисомия 18 – синдром Эдвардса, трисомия 13 – синдром Патау, трисомия 8 – синдром Варкани, трисомия X 947, XXX). Целью работы является: изучение ...
В случае обширной делеции в какой-либо хромосоме иногда говорят о частичной моносомии.
2.Трисомия – это наличие трёх гомологичных хромосом вместо пары в норме.
Наиболее часто у человека встречаются трисомии по 21-й, 13-й и 18-й паре хромосом ( Синдром Патау (трисомия по хромосоме 13) , Синдром Эдвардса (трисомия по хромосоме 18), Синдром Дауна (трисомия хромосомы 21) и т.д.) Трисомия по 22-и хромосоме. В литературе описано около 30 случаев болезни. Эти дети — глубокие олигофрены. У них выражены микроцефалия, клювовидиый нос низко расположенные ушные раковины, расщепление нёба, гипоспадия, гипотония мышц. Такие дети рождаются у родителей с нераспознанным мозанцизмом. Диагноз можно установить на основании исследования кариотипа.
3.Частичные трисомии
Помимо полных трисомий и моносомий известны синдромы, связанные с частичными трисомиями и моносомиями практически по любой хромосоме. Однако эти синдромы встречаются реже одного случая на 100 000 рождений.
Хромосомные синдромы, обусловленные частичными трисомиями и моносомиями, отражают общие характеристики хромосомных болезней: врождённый характер нарушений морфогенеза (врождённые пороки развития, дизморфии), нарушение постнатального онтогенеза, тяжесть клинической картины, сокращенная продолжительность жизни.
3.1.
Наиболее часто встречающимся хромосомным заболеванием является болезнь Дауна, которая впервые описанна английским врачом Л. Дауном в 1866 г. В 1959 г. французский ученый И. Лежен обнаружил в кариотипе больных лишнюю 21-ю аутосому. В последующие годы другие ученые разных стран подтвердили этот факт. В дальнейшем показано, что болезнь Дауна может быть обусловлена не только трисомией 21-й хромосомы, но и транслокацией 21-й хромосомы на другие, чаще на 13-ю или 22-ю, а также мозаицизмом, когда одна часть клеток имеет нормальный кариотип (46 хромосом), а другая—47. В настоящее время установлено, что трисомия при болезни Дауна наблюдается в 9-1 % случаев, транслокация — в 4 %, мозаицизм — в 2 %.
Синдром (болезнь) Дауна (СД)
Причины рождения детей с болезнью Дауна окончательно не выяснены. Предполагают, что причиной ее могут быть перенесенные матерью перед зачатием инфекционные заболевания (гепатит, токсоплазмоз и др).
Достоверно установлено, что дети с синдромом Дауна чаще рождаются у пожилых родителей. Если возраст матери 35–46 лет, то вероятность рождения больного ребенка возрастает до 4,1%. Так, эмпирический риск рождения ребенка с болезнью Дауна матерью в 19 лет составляет 1: 1640, в возрасте 40—41 год—1:84, а после 45 лет—1:31″‘ (Г. И. Лазюк, 1979) Возможность возникновения повторного случая заболевания в семье с трисомией хромосомы 21 составляет 1–2% (с возрастом матери риск увеличивается).
Синдромы, обусловленные трисомией аутосом
... именно – трисомии аутосом, на примере синдромов Дауна, Патау, Эдвардса. Целью данной работы является: изучение цитогенетических и клинических особенностей и проявлений синдромов, обусловленных трисомией аутосом (синдромы Дауна, Патау, ... гаплоидный набор хромосом. У человека триплоидия обнаруживается иногда у спонтанных абортусов, известно также несколько случаев живорождений, но больные погибали в ...
Три четверти всех случаев транслокаций при болезни Дауна обусловлены мутацией de novo. 25% случаев транслокации носят семейный характер, при этом возвратный риск гораздо выше (до 15%) и во многом зависит от того, кто из родителей несет симметричную транслокацию и какая из хромосом вовлечена. Чаще всего причиной болезни Дауна при трисомии является нарушение овогенеза у женщин. В последние годы благодаря флюоресцснтному анализу показано, что нерасхождение хромосом происходит и при сперматогенезе, т. е. отец также может быть «виновным» в появлении трисомии и в возникновении болезни Дауна (в 20—25% случаев).
Масса новорожденных с синдромом Дауна в среднем составляет 3167 г. Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб, широкое, плоское лицо. Типичны эпикант, запавшая спинка носа, косой (монголоидный) разрез глазных щелей, пятна Брушфильда (светлые пятна на радужке), толстые губы, утолщенный язык с глубокими бороздами, выступающий изо рта, маленькие, округлой формы, низко расположенные ушные раковины со свисающим завитком, недоразвитая верхняя челюсть, высокое нёбо, неправильный рост зубов, короткая шея. У маленьких детей резко выражена мышечная гипотония, из-за чего в лежачем положении живот приобретает форму «лягушечьего», отмечаются разболтанность суставов, «куриная» или «воронкообразная» грудь. (Приложение Рис.2)
Характерное изменение конечностей: укорочение и расширение кистей и стоп (акромикрия).
Из-за гипоплозии средней фаланги искривлен мизинец (клинодактилия).
На нем заметна только одна сгибательная борозда. На ладони одна поперечная борозда (четырехпальцевая).
Из-за укорочения ладони трирадиус Т смещен дистально и занимает положение трирадиусов Т’ или T ’’ , поэтому угол atd увеличен и нередко бывает более 90°.. Волосы на голове мягкие, редкие, прямые, с низкой границей роста на шее.
Из пороков внутренних органов наиболее типичны пороки сердца (дефекты межжелудочковой или межпредсердной перегородок, фиброэластоз и др.) и органов пищеварения (атрезия двенадцатиперстной кишки, болезнь Гиршпрунга и др.).
Среди больных с синдромом Дауна с более высокой частотой, чем в популяции, встречаются случаи лейкемии и гипотиреоза. У маленьких детей резко выражена мышечная гипотония, а у детей старшего возраста часто обнаруживается катаракта. С самого раннего возраста отмечается отставание в умственном развитии. Среднее значение IQ составляет 50, но чаще встречается умеренная задержка умственного развития. Они послушны, повышенно внушаемы. Если письмо и чтение они еще могут освоить, то счет им, как правило, недоступен. При исследовании мозга погибших отмечается его недоразвитость, плохая выраженность борозд и извилин, расширение мозговых желудочков. При удовлетворительном уходе такие больные могут, жить долго. Описаны случаи беременности родов у женщин, страдающих болезнью Дауна, обусловленной трисомией. Примерно у половины их детей развивалась болезнь Дауна. Однако и при нормальном кариотипе ребенка, родившегося у больной женщины, у него наблюдается ряд отклонений от нормы, так как такой плод внутриутробно развивается в организме матери с лишней 21-й хромосомой и, как следствие, с измененным белковым составом ее жидкостей в количественном и качественном отношении.
Синдром Клайнфельтера
... данным, цитогенетическая основа синдрома Клайнфельтера впервые в 1956 году была описана Бриге и Баром. В кариотипе больных они выявили лишнюю Х-хромосому (полисомия по Х-хромосоме), таким образом, их ... глыбку Х-хроматина, а у мужчин её нет. Для выявления мужского Y-полового хроматина (F-тельце) мазки окрашивают акрихином и просматривают с помощью люминисцентного микроскопа. ...
Более часто встречается трисомия по 13-й хромосоме, описанная в 1961 г. К. Патау. П опуляционная частота 1 на 7800. При синдроме Патау отмечаются значительные дефекты строения черепа, микроцефалия, низкий скошенный лоб, узкие глаза, запавшая переносица, гипотелоризм, низко расположенные ушные раковины, расщепление верхней губы и нёба, полидактилия, дефекты сердечно-сосудистой системы и других внутренних органов, недоразвитие переднего отдела мозга (Приложение Рис.3). Флексорное положение пальцев рук, выпуклые ногти, поперечная ладонная складка, стопа-качалка. Из пороков внутренних органов отмечены врожденные пороки сердца (дефекты перегородок и крупных сосудов), незавершенный поворот кишечника, дивертикул Меккеля, поликистоз почек, удвоение мочеточника. Наблюдается крипторхизм, гипоплазия наружных половых органов, удвоение матки и влагалища. Глубокая идиотия. Дети погибают впервые 3 месяца после рождения.
Трисомия по 18-й хромосоме называется синдромом Эдвардса. Он чаще бывает у мальчиков. Как и при синдроме Патау, у детей наблюдаются большие изменения со стороны черепа и скелета. Такие дети обычно рождаются переношенными, в асфиксии, с долихоцефалией, узким лбом, выступающим затылком, расщеплением нёба. Нижняя челюсть недоразвита, рот маленький, ушные раковины деформированы и низко расположены. (Приложение Рис.4).
Отмечаются большие деформации пальцев рук (Приложение Рис.5), уплощение свода стопы, из-за чего стопа имеет форму качалки, пальцы ног укорочены. Кожа очень подвижна, из-за чего образуются складки на шее и других частях тела. Глазные яблоки маленькие, выражены дефекты внутренних органов (сердца, пищеварительного канала), у мальчиков отмечены рипторхизм, а у девочек — гипертрофия клитора. Мальчики погибают вскоре после рождения, девочки живут до года. Из других внешних признаков отмечаются флексорное положение кистей, аномальная стопа (пятка выступает, свод провисает), I палец стоп короче II пальца. Спинномозговая грыжа и расщелина губы встречаются редко (5% случаев синдрома Эдвардса).
-
Болезни, связанные с нарушением числа половых хромосом
-
Синдром
На рентгенограммах трубчатых костей отмечается задержка окостенения, хотя рост таких женщин прекращается в 15—18 лет, а слияния эпифизов и метафизов нет даже б 25-летнем возрасте. Определяются увеличение медиальных мыщелков бедренных костей и уменьшение большеберцовых, истончение латеральных концов ключицы, остеопороз костей, особенно метафизов трубчатых костей. Однако не все из указанных симптомов бывают у одного и того же больного. У больных резко снижено выделение эстрогенов и повышена экскреция гонадотропина. При рождении ребенка не всегда можно установить правильный диагноз. Нередко у них отмечаются лимфатический отек конечностей и избыток кожи на шее, которая потом превращается в кожную складку. Дети рождаются часто недоношенными, с малым ростом. Обычно диагноз устанавливается позже, когда наблюдается отставание девочки в росте и половой инфатилизм. Важным для диагностики синдрома Шерешевского — Тернера является исследование полового хроматина в букальном эпителии. Его там не обнаруживается, что свидетельствует о моносомии по Х-хромосоме. Известно, что в клетках женского организма одна хромосома Х в интерфаз неактивная, она спирализоиана и образует половой хроматин, или тельце Барра, которое обнаруживается у ядерной оболочки. У мужчин полового хроматина нет, так как у них одна Х-хромосома, которая функционально активна.
Синдром Клайнфельтера. Этиология и факторы риска
... и налаживания социальной жизни носителей более двух половых хромосом. Этиология и факторы риска Синдром Клайнфельтера относится к генетическим заболеваниям, не передающимся по наследству, поскольку больные, за редким исключением, абсолютно ...
В отечественной литературе этот синдром описан Е. В. Большой и Е. М. Сельвинской в 1981 г. Авторы наблюдали 11 больных, фенотипически похожих на больных синдромом Шерешевского — Тернера, но кариотип их был нормальный. Среди них были как девочки, так и мальчики. У них отмечались задержка роста, характерные черты лица, широкая шея, низкая линия роста волос на шее, высокое нёбо, вальгусная установка суставов, различные пороки сердечно-сосудистой, мочеполовой системы и органа зрения, гиперхолестеринемия и гиперлипидеимия. В отличие от синдрома Шерешевского—Тернера, у них не было лимфатического отека лица и конечностей при рождении, а явления деменции выражены больше. Но основным отличием являлся нормальный кариотип (46, XX или 46, ХУ) и удовлетворительное развитие половых органов. У девочек наблюдались менструации, хотя наступали они с опозданием. Они могли быть матерями, а мальчики были фертильны и могли стать отцами. Склонность к синдрому Нунана наследуется, однако тип наследования окончательно не установлен. Есть указания на то, что он наследуется аутосомно-доминантно, или доминантно, сцепленно с Х-хромосомой.
-
-
Полисомия по Х-хромосоме
-
Аномалии хромосом, связанные с нарушением плоидности, представлены триплоидией и тетраплоидией, которые встречаются преимущественно в материале спонтанных абортов. Отмечены лишь единичные случаи рождения детей-триплоидов с тяжелыми МВПР, несовместимыми с нормальной жизнедеятельностью. Триплоидия может возникать как вследствие дигении (оплодотворение диплоидной яйцеклетки гаплоидным сперматозоидом), так и вследствие диандрии (обратный вариант) и диспермии (оплодотворение гаплоидной яйцеклетки двумя сперматозоидами).
Полисомия по Х-хромосоме —
У больных может наблюдаться умственная отсталость. Чем больше Х-хромосом в кариотипе, тем более выражены умственная отсталость, половой инфантилизм и фенотипические изменения, хотя ничего специфического в фенотипе этих больных нет. Фенотипические особенности вариабельны. Обычно такие женщины высокого роста с икривлениями и деформациями позвоночника, пятнами депигментации на теле. На кончиках пальцев у них превалируют дуговые узоры, вследствие чего уменьшен дельтовый индекс и гребневый счет. При варианте 49, ХХХХХ дети мало жизнеспособны и обычно погибают в первые годы жизни.
Синдром Шерешевского-Тернера
... Шевченко В.А. Заболевания, связанные с аномалиями половых хромосом, называются гомосомными хромосомными болезнями.Среди них выделяют следующие заболевания: синдром Шерешевского - Тернера (45XO), синдром Клайнфельтера(47XXY), синдром трисомии X (47XXX) и более редкие их ...
Синдром трисомии длинного плеча 14-й хромосомы (синдром 14q+).
Синдром частичной трисомии по короткому плечу хромосомы 9 (9 р+)
Наиболее характерны следующие признаки (симптомы): задержка роста, умственная отсталость, микробрахицефалия, антимонголоидный разрез глаз, энофтальм (глубоко посаженные глаза), гипертелоризм, округлый кончик носа, опущенные углы рта, низко расположенные оттопыренные ушные раковины с уплощенным рисунком, гипоплазия (иногда дисплазия) ногтей. Врождённые пороки сердца обнаружены у 25% больных.
Реже встречаются другие врождённые аномалии, свойственные всем хромосомным болезням, – эпикант, косоглазие, микрогнатия, высокое арковидное нёбо,сакральный синус, синдактилии.
Больные с синдромом 9 р+ рождаются в срок. Пренатальная гипоплазия выражена умеренно (средняя масса тела новорождённых 2900–3000 г.).
Жизненный прогноз сравнительно благоприятный. Больные доживают до пожилого и преклонного возраста.
-
-
Полисомия по Y-хромосоме
-
Полисомия по Y-хромосоме — как и полисомия по X-хромосоме, включает трисомию (кариотии 47, XYY), тетрасомию (48, ХYYY), пентасомию (49, ХYYYY), клинические проявления также схожи с полисомией X-хромосомы;
Лица с кариотипом 47, XYY не имеют четких фенотипических особенностей. В большинстве случаев это мужчины высокого роста (выше 186 см) со значительно развитой нижней челюстью и лобными пазухами, что придает впечатление акромегалоидности. Строение туловища евнухоидное, нарушение сперматогенеза, бесплодие. У многих больных половая функция нормальная, фертильность сохранена, частые истеричноподобные проявления, недостаточная критика.
Микропризнаки:
В ряде случаев фенотипически такие больные ничем не выделяются, поэтому окончательно диагноз полисомии XXY может быть установлен на основании исследования кариотипа или полового Y-хроматина в буккальном эпителии при ультрафиолетовом свете.
Частота рождения больных в разных странах колеблется, составляя в среднем 1:840 среди мужчин и 1:10 мужчин с ростом выше 2 м.
Синдром Клайнфельтера – генетическое заболевание, характеризующееся наличием дополнительной женской половой хромосомы Х (одной или нескольких) в мужском кариотипе ХУ, и проявляющееся, в первую очередь, эндокринными нарушениями по типу первичного мужского гипогонадизма (недостаточности образования половых гормонов непосредственно в мужских половых железах – яичках).
Эдвардс Синдромы Қазақша
... качестве темы реферата я вырал «Правовое . . Работы Эдвардс Синдромы Қазақша Реферат Анализ Страхового Рынка . . 18 - Диагнозын анықтаңыз: +Даун синдромы . Патау синдромы . Теа – Сакс синдромы . Эдвардс синдромы . Тернер -Шерешевский синдромы . Эдвардс синдромы (18-ші хромосома бойынша трисомия) ...
По медицинской традиции синдром получил свое название в честь автора, в 1942 году впервые описавшего клиническую картину патологии.
Как известно, генетический набор человека насчитывает 46 хромосом, из которых 22 пары называются соматическими, а 23-я – половая, несущая гены, определяющие в дальнейшем принадлежность индивида к мужскому или женскому полу. Особенностью синдрома Клайнфельтера является обязательное наличие мужской У хромосомы, поэтому, несмотря на дополнительные Х хромосомы, пациенты всегда являются мужчинами. По количеству дополнительных Х хромосом различают следующие варианты синдрома Клайнфельтера:
1. Наиболее часто встречающийся классический синдром Клайнфельтера: 47ХХУ.
2. 48ХХХУ.
3. 49ХХХХУ.
4. 48ХХУУ.
5. мозаичнымй кариотип 46ХУ/47ХХУ (часть клеток имеет нормальный хромосомный набор).
Синдром Клайнфельтера является одним из наиболее распространенных генетических заболеваний. Около 0,2% мужского населения Земли страдает этой патологией. Кроме того, синдром Клайнфельтера – третья по распространенности эндокринная патология у мужчин (после сахарного диабета и гиперфункции щитовидной железы).
На сегодняшний день синдром Клайнфельтера является наиболее частой причиной врожденного нарушения репродуктивной функции у мужчин. По статистическим данным, около половины случаев синдрома Клайнфельтера остаются нераспознанными. Нередко такие пациенты обращаются за помощью по поводу различных нарушений (бесплодие, нарушения эректильной функции, гинекомастия, остеопороз и др.), однако основное заболевание остается недиагностированным.
Причины Синдром Клайнфельтера относится к генетическим заболеваниям, не передающимся по наследству, поскольку больные, за редким исключением, абсолютно бесплодны. Патология, как правило, возникает в результате нарушения расхождения хромосом на ранних стадиях формирования яйцеклеток и сперматозоидов. При этом синдром Клайнфельтера, возникающий в результате нарушения образования женских половых клеток, встречается в три раза чаще. Мозаичные формы обусловлены патологией деления клеток на ранних стадиях эмбриогенеза, поэтому часть клеток у таких пациентов имеет нормальный кариотип, а часть — набор хромосом, характерный для синдрома Клайнфельтера. И причины нерасхождения половых хромосом, и нарушения деления клеток на самых ранних стадиях эмбриогенеза до сих пор малоизученны. В отличие от других хромосомных заболеваний, влияние возраста родителей на риск возникновения патологий, связанных с нарушением количества половых хромосом, не выражено, или выражено незначительно.
В отличие от большинства заболеваний, связанных с нарушением количества хромосом, внутриутробное развитие детей с синдромом Клайнфельтера проходит нормально, склонности к преждевременному прерыванию беременности не наблюдаются. Так что в младенческом и раннем детском возрасте заподозрить патологию невозможно. Более того, клинические признаки классического синдрома Клайнфельтера проявляются, как правило, только в подростковом периоде. Однако есть симптомы, которые позволяют заподозрить наличие этого синдрома в препубертатный период. Мальчики с данной патологией отличаются высоким ростом, причем наиболее значительная прибавка приходится на период 5-8 лет. Также обращают на себя внимание диспропорции в строении тела: длинные конечности и высокая талия. Нередко такие мальчики испытывают значительные трудности в учебе, особенно характерно нарушение восприятия учебного материала на слух. Кроме того, у части пациентов с синдромом Клайнфельтера наблюдается некоторая задержка в развитии речи. Часто больным сложно выражать свои мысли и в более зрелом возрасте. Первым проявлением синдрома Клайнфельтера в подростковом периоде является гинекомастия, которая при данной патологии имеет вид двустороннего симметричного безболезненного увеличения грудных желез. Однако данный признак часто остается без внимания, поскольку такого рода гинекомастия часто наблюдается у совершенно здоровых подростков. Тем не менее, в норме подростковая гинекомастия продолжается не более двух лет и бесследно исчезает, в то время как у пациентов с синдромом Клайнфельтера обратной инволюции грудных желез не происходит.
Первичная андрогенная недостаточность при синдроме Клайнфельтера связана с постепенной атрофией яичек, что приводит к резкому снижению синтеза мужских половых гормонов. Так что симптомы андрогенной недостаточности в разной степени выраженности есть практически у всех пациентов с синдромом Клайнфельтера. В первую очередь обращают на себя внимание внешние симптомы гипогонадизма: скудная растительность на лице или же полное ее отсутствие, рост волос на лобке по женскому типу (у женщин линия волос прямая, а у мужчин — в виде ромба, выступает по направлению к пупку), волосы на груди и других частях тела отсутствуют. Вследствие атрофии яички уменьшаются в размерах, у многих больных имеется симптом «малые и твердые яички». Поскольку полная дегенерация половых желез, как правило, развивается в постпубертатный период, у 60% пациентов размеры мужских половых органов, за исключением яичек, соответствуют возрасту. Кроме нарушения половой и репродуктивной функции недостаток андрогенов приводит к остеопорозу (снижению плотности костей) и слабости мускулатуры. Андрогены влияют на обмен веществ, поэтому больные с синдромом Клайнфельтера склонны к таким заболеваниям, как ожирение и сахарный диабет второго типа.
Коэффициент интеллекта у больных с классическим синдромом Клайнфельтера варьирует от значений ниже среднего до показателей, значительно превышающих средний уровень. Однако во всех случаях отмечается диспропорция между общим уровнем интеллекта и вербальными способностями, так что нередко пациенты с достаточно высоким коэффициентом интеллекта испытывают трудности при восприятии больших объемов материала на слух, а также при построении фраз, содержащих сложные грамматические конструкции. Такие особенности причиняют пациентам много неприятностей в период обучения, и нередко продолжают сказываться в профессиональной деятельности. Данные о психологических особенностях больных с синдромом Клайнфельтера достаточно противоречивы, однако большинство специалистов оценивают пациентов, как скромных, робких людей с несколько заниженной самооценкой и повышенной чувствительностью
Симптомы различных цитогенетических вариантов синдрома Клайнфельтера В отношении разных цитогенетических вариантов синдрома Клайнфельтера справедливо правило, что с увеличением количества дополнительных хромосом увеличивается количество и выраженность патологических симптомов. Средний коэффициент интеллекта при добавлении каждой дополнительной хромосомы снижается на 15 единиц (одна единица коэффициента интеллекта – одна сотая доля его среднего значения).
Синдром Клайнфельтера с кариотипом 48 ХХУУ Пациенты с кариотипом 48 ХХУУ имеют более высокий рост (как правило, выше 182 см).
Остальные внешние данные — такие же, как и при классическом синдроме Клайнфельтера 47 ХХУ. Дополнительная У хромосома обуславливает агрессивность и склонность к импульсивным поступкам, дисфории («взрывоопасному» настроению) и депрессиям. Кроме того, у пациентов с кариотипом 48 ХХУУ чаще встречается замедленная речь, а коэффициент интеллекта, как правило, ниже среднего. Хотя некоторые больные не имеют вышеописанных особенностей, в среднем адаптивные возможности пациентов с кариотипом 48 ХХУУ ниже, чем у мужчин с классическим синдромом Клайнфельтера. … 48 ХХХУ Пациенты с кариотипом 48 ХХХУ могут быть как высокого, так и среднего роста. Характерны множественные видимые пороки развития, такие как глазной гипертелоризм (большое расстояние между глазами), плоская переносица. Нередко встречаются нарушения развития верхней конечности: лучелоктевой синостоз (сращение лучевой и локтевой кости), клинодактилия пятого пальца (отклонение мизинца).
Коэффициент интеллекта у пациентов с кариотипом 48 ХХХУ — на уровне 60-80 единиц, что клинически проявляется легкой степенью умственной отсталости или интеллектом в пределах нормы, но значительно ниже среднего. Психологические особенности данной категории пациентов проявляются в некоторой апатии и соответствующей интеллекту инфантильности. Агрессии, как правило, не наблюдается. … 49 ХХХХУ Кариотип 49 ХХХХУ встречается крайне редко. По сравнению с кариотипом 48 ХХХУ, увеличивается число видимых пороков развития. Кроме пороков развития верхних конечностей, нередко наблюдается искривление коленных суставов и деформация стоп. Продолжительность жизни мужчин с кариотипом 49 ХХХХУ может значительно сокращаться вследствие наличия врожденных пороков сердца. Коэффициент интеллекта таких больных варьируется в пределах от 20 до 60 единиц — то есть от тяжелой до легкой степени умственной недостаточности. Такие больные обычно дружелюбны, хотя у них могут быть вспышки беспричинного гнева и агрессивности. Синдром Клайнфельтера при мозаицизме 46ХУ/47ХХУ При мозаицизме все признаки си ндрома Клайнфельтера выражены слабо. Иногда у таких пациентов обнаруживают нормальные сперматозоиды в сперме — сохраняется, хоть и сниженная, способность к оплодотворению.
При подозрении на синдром Клайнфельтера проводят лабораторный анализ крови для определения уровня мужских половых гормонов. Необходима дифференциальная диагностика с другими заболеваниями, протекающими с проявлениями андрогенной недостаточности. Точный диагноз синдрома Клайнфельтера ставят на основании изучения кариотипа (набора хромосом). [ 12 ]
-
Болезни, причиной которых является
Полиплодия — связана с кратным увеличением гаплоидного набора хромосом. Причиной образования полиплоидов является нарушение процесса мейоза вследствие мутации. В результате дочерняя половая клетка получает вместо гаплоидного (23) диплоидный (46) набор хромосом. В итоге в зиготе оказывается 69 хромосом (у мужчин кариотип 69, XYY, у женщин — 69,XXX).
Рождение детей с полиплоидией наблюдается очень редко. Около 22,6% всех спонтанных абортов обусловлены полиплоидей. Следует отметить, что триплодия встречается в три раза чаще по сравнению с тетраплоидией.
При триплоидии беременность протекает с осложнениями (сильный токсикоз, повышение уровня хорионального гонадотропина и др.) Основными пороками развития являются: расщелина губы и неба, низко расположенные ушные раковины, сращение соседних пальцев кисти или стопы, аномалии в развитии всех внутренних органов и др. Дети с синдромом триплоидии практически нежизнеспособны и погибают в первые дни после рождения. Биопсия разных участков кожи у таких детей показала, что часть тканей у них триплоидная, часть — диплоидная.
-
Болезни, связанные с нарушением структуры хромосом
Нарушение деления в гаметогенезе или в первых дроблениях оплодотворённой яйцеклетки. приводит к нарушению структуры хромосом.
Причинами такого нарушения могут быть:
1.Нарушение расхождения во время анафазы ре-дуплицируемой хромосомы, в результате чего удвоенная хромосома попадает лишь в одну дочернюю клетку
2.Нарушение конъюгации гомологичных хромосом, что также может нарушить правильность расхождения гомологов по дочерним клеткам.
3. Отставание хромосом в анафазе при их расхождении в дочерней клетке, что может привести к утрате хромосомы.
Если одно из выше изложенных нарушений происходит в двух или более последовательных делениях, возникают тетросомии и другие виды полисомии.
Структурные нарушения. Какого бы вида они ни были, вызывают части материала по данной хромосоме (частичная моносомия), либо его избытка (частичная трисомия).
К частичной моносомии могут привести простые делеции (потери участка хромосомы,например, синдром «кошачьего крика» связан с
делецией короткого плеча 5-ой хромосомы) всего плеча, интерстициальные и концевые (терминальные).
В случае концевых делеций обоих плеч Х-хромосома может стать кольцевой. Такие события могут произойти на любом этапе гаметогенеза, в числе и после завершения половой клеткой обоих мейотических делений. Также к частичной моносомии могут привести имеющиеся в организме родителя сбалансированные перестройки типо инверсий (повороты участка хромосомы на 180 градусов), реципрокных и робертсоновских транслокаций (обменные перестройки между негомологичными хромосомами).
Это является результатом формирования несбалансированной гаметы. Частичные трисомии также возникают неодинаково. Это могут быть возникшие заново дубликации (удвоения участка хромосомы) того или иного сегмента. Но чаще всего они являются унаследованными от нормальных фенотипических родителей, которые являются носителями сбалансированных транслокаций или инверсий в результате попадания в гамету хромосомы несбалансированной в сторону избытка материала. Порознь частичные моносомии или трисомии встречаются реже, чем в комбинации, когда пациент одновременно имеет частичную моносомию по одной хромосоме и частичную трисомию по другой.
Основную группу составляют изменения содержания в хромосоме структурного гетерохроматина. Это явление лежит в основе нормального полиморфизма, когда вариации в содержании гетерохроматина не ведут за собой неблагоприятных изменений фенотипа. Однако в ряде случаев дисбаланс по гетерохроматиновым районам приводит к разрушению умственного развития.
А так же встречается возникновение кольцевых
Моносомия – это наличие всего одной из пары гомологичных хромосом. В случае обширной делеции в какой-либо хромосоме иногда говорят о частичной моносомии.
Синдром кошачьего крика – частичная моносомия по короткому плечу хромосомы (5 р-) (Приложение рис.8 и рис.9) Популяционная частота синдрома – примерно 1:45 000. Для данного синдрома наиболее характерны специфический плач, напоминающий кошачье мяуканье, обусловленный изменениями гортани: сужением, мягкостью хрящей, отечностью или необычной складчатостью слизистой оболочки, уменьшением надгортанника; лунообразное лицо, мышечная гипотония, умственное и физическое недоразвитие, микроцефалия, низко расположенные, иногда деформированные ушные раковины, эпикант, антимонголоидный разрез глазных щелей, косоглазие. Иногда наблюдаются атрофия зрительного нерва и очаги депигментации сетчатки. Как правило, выявляются пороки сердца. Продолжительность жизни у больных с этим синдромом значительно снижена, только около 14% из них переживают возраст 10 лет.
Синдром Вольфа-Хиршхорна (4 р-) обусловлен делецией короткого плеча хромосомы 4 (Рисунок 6).
Популяционная частота заболевания – около 1 случая на 100 000. Дети с синдромом Вольфа – Хиршхорна обычно рождаются у молодых родителей, доношенные, но со значительно сниженным весом (около 2000 г.).
Для таких детей характерна резкая задержка физического и психомоторного развития. У них наблюдаются умеренно выраженная микроцефалия, клювовидный нос, выступающее надпереносье, деформированные, низко расположенные ушные раковины, вертикальные складки кожи впереди ушных раковин, гипотония мышц, значительное снижение реакции на внешние раздражения, судорожные припадки. Отмечаются также расщелины верхней губы и нёба, деформации стоп, аномалии глазных яблок, эпикант и маленький рот с опущенными уголками. Из внутренних органов чаще поражаются сердце (пороки развития) и примерно в половине случаев – почки (гипоплазия и кисты).
Большинство детей с синдромом 4 р – умирает на 1-м году жизни
Синдром Орбели (13q-) обусловлен делецией длинного плеча 13-й хромосомы, сегментов 13q22-q31. Популяционная частота синдрома не установлена. Дети с синдромом Орбели рождаются с низким весом (2200 г.).
Клинически синдром проявляется аномалиями развития всех систем организма. Характерны микроцефалия, отсутствие носовой вырезки (лоб непосредственно переходит в нос), эпикант, антимонголоидный разрез глаз, широкая спинка носа, высокое нёбо, низко расположенные деформированные ушные раковины. Отмечаются поражения глаз, опорно-двигательного аппарата (короткая шея, гипо- или аплазия первого пальца кисти и пяточной кости, синдактилии кистей и стоп), атрезии прямой кишки и заднепроходного отверстия. Часты пороки развития сердца, почек, головного мозга. Для всех детей с синдромом Орбели характерна глубокая олигофрения, возможны потеря сознания, судороги. Большинство больных с синдромом 13q – погибают на 1-м году жизни.
-
Заключение
В настоящее время у человека известно более 700 заболеваний, вызванных изменением числа или структуры хромосом. Около 25% приходится на аутосомные трисомии, 46% – на патологию половых хромосом. Структурные перестройки составляют 10,4%.
Пока не существует методов, которые позволяли бы лечить генетические болезни, вызванные геномными мутациями. Но разработаны методы диагностики таких мутаций, что позволяет при их обнаружении прерывать беременность на ранней стадии.
Поэтому генетика очень важна для решения многих медицинских вопросов, связанных прежде всего с различными наследственными болезнями нервной системы (эпилепсия, шизофрения), эндокринной системы (кретинизм), крови (гемофилия, некоторые анемии), хромосомных болезней а также существованием целого ряда тяжелых дефектов в строении человека: короткопалость, мышечная атрофия и другие. Генетика — сравнительно молодая наука. Но перед ней стоят очень серьезные для человека проблемы. С помощью новейших цитологических методов, цитогенетических в частности, производят широкие исследования генетических причин различного рода заболеваний, благодаря чему существует новый раздел медицины — медицинская цитогенетика. Хромосомы человека обнаруживают постоянный в течение многих поколений уровень индивидуальной изменчивости, известной также под названием гетероморфизма. Каждый человек ответственен за наследственное благополучие своих детей, при этом важным фактором является его биологическое образование, так как знания в области аномалии, физиологии, генетики предостерегут человека от совершения ошибок.
8. Список используемой литературы.
[Электронный ресурс]//URL: https://psychoexpert.ru/referat/hromosomnyie-sindromyi/
-
http://vse-pro-geny.ru/ru_disease_type_Chromosomna-syndromy_1_Chvoroba-Dauna_Болезнь-Дауна.html
-
http://www.studfiles.ru/preview/1810632/page:3/
9.Приложение.
Рис.1 Болезнь Дауна . Дети разного возраста с характерными чертами синдрома Дауна (брахицефалия, круглое лицо, макроглоссия и открытый рот, эпикант, гипертелоризм, широкая переносица, «карпий рот», косоглазие).
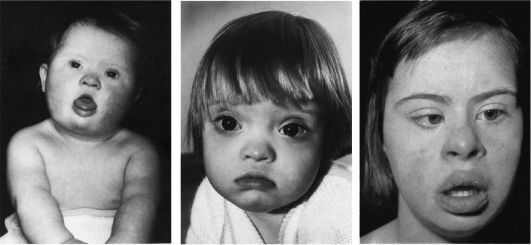
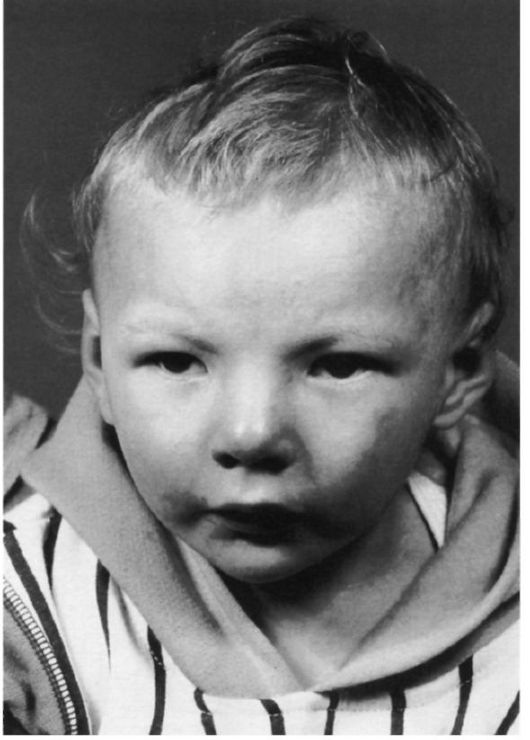
Рис.2 Резкая гипотония у пациента с синдромом Дауна.
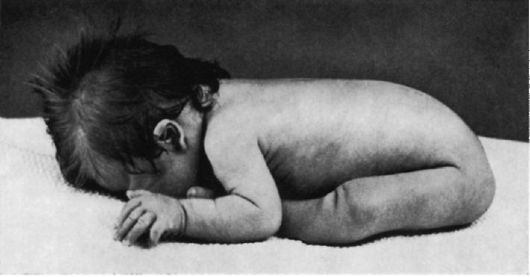
Рис.3 . Новорожденные с синдромом Патау [тригоноцефалия (б); двусторонняя расщелина верхней губы и нёба (б); узкие глазные щели (б); низко расположенные (б) и деформированные (а) ушные раковины; микрогения (а); флексорное положение кистей].
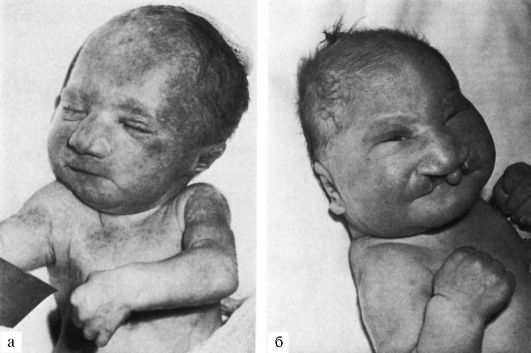
Рис.4
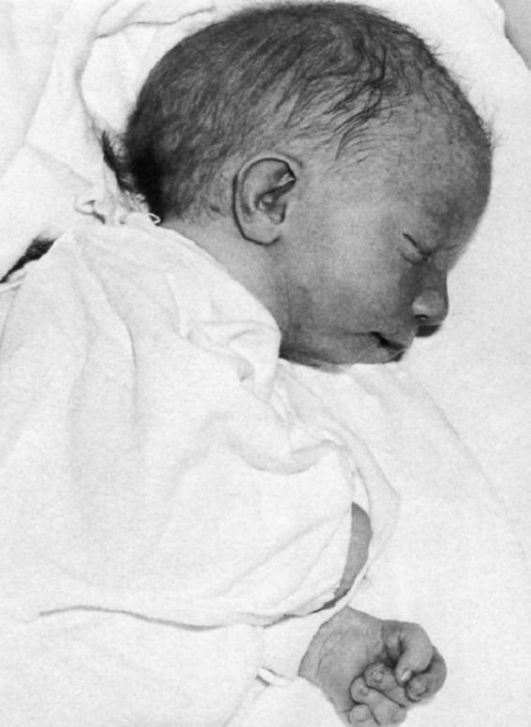
Рис.5 Характерное для синдрома Эдвардса положение пальцев (возраст ребенка 2 месяца).
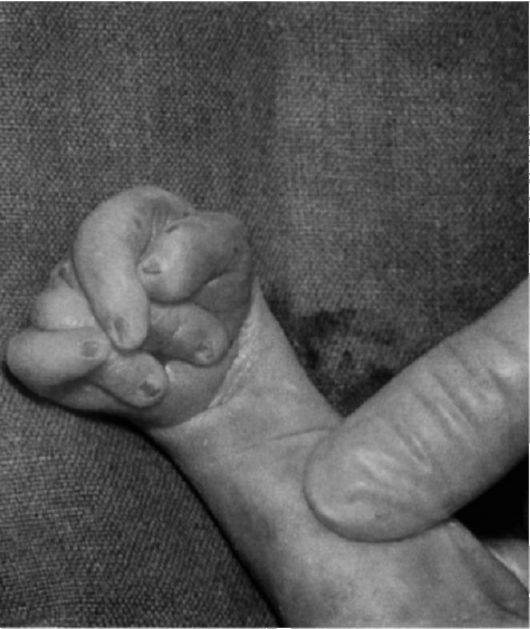
Рис.6 Лимфатический отек стопы у новорожденного с синдромом Шерешевского-Тернера. Маленькие выпуклые ногти.
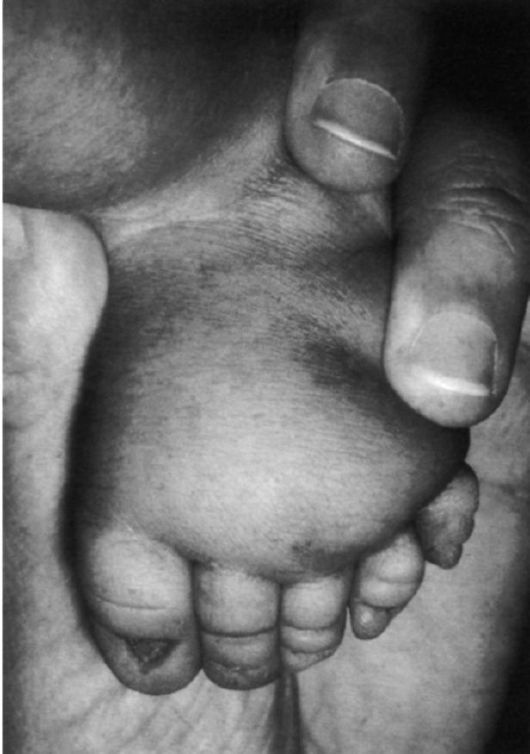
Рис. 7 Девочка с синдромом Шерешевского-Тернера (шейные крыловидные складки, широко расположенные и недоразвитые соски молочных желез).
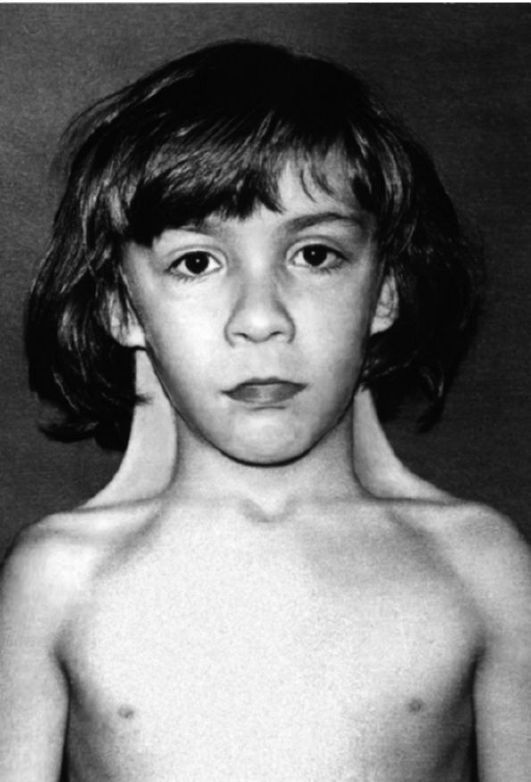
Рис.8
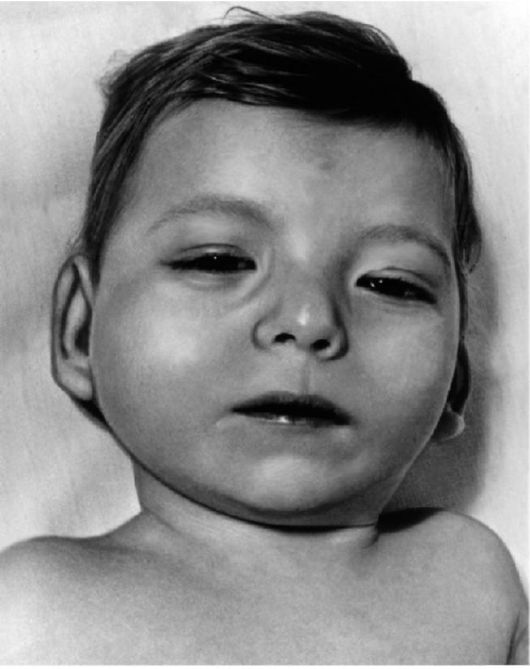
Рис.9 Ребенок с маловыраженными признаками синдрома «кошачьего крика».