
Наследственные заболевания – заболевания обусловленные изменениями (мутациями), преимущественно хромосомными или генными. Соответственно чему условно выделяют хромосомные и собственно наследственные (генные) болезни. Известно около 3 тыс. наследственных болезней и синдромов, многие являются причиной высокой детской смертности. В профилактике наследственных болезней важную роль играет медико-генетическое консультирование.
Наследственные болезни известны человечеству, по-видимому, со времен Гиппократа, однако их изучение началось лишь в 20 веке после переоткрытия законов Менделя. На протяжении первых десятилетий 20 века происходило накопление и анализ фактических данных по наследованию патологических признаков. Дальнейшее понимание природы наследственных болезней связано с успехами в изучении механизма реализации генетической информации.
По мере развития медицины возможность выявления наследственных заболеваний увеличивается. Этот фактор указывает на растущее значение медицинской генетики и генетики человека. Меры, принятые при раннем выявлении наследственных болезней, могут предотвратить их развитие.
В данной работе рассматриваются численные хромосомные мутации, а именно – трисомии аутосом, на примере синдромов Дауна, Патау, Эдвардса.
Целью данной работы является: изучение цитогенетических и клинических особенностей и проявлений синдромов, обусловленных трисомией аутосом (синдромы Дауна, Патау, Эдвардса).
ГЛАВА 1. ХРОМОСОМНЫЕ МУТАЦИИ. ТИПЫ И ПРИЧИНЫ ВОЗНИКНОВЕНИЯ ХРОМОСОМНЫХ МУТАЦИЙ
1.1 Численные и структурне хромосомне мутации
Различают два основных типа хромосомных мутаций: численные хромосомные мутации и структурные хромосомные мутации. В свою очередь численные мутации делятся на ан- эуплоидии, когда мутации выражаются в утрате или появлении дополнительной одной либо нескольких хромосом, и полиплоидии, когда увеличивается число гаплоидных наборов хромосом. Потерю одной из хромосом называют моносомией, а возникновение дополнительного гомолога у любой пары хромосом — трисомией. Структурные хромосомные мутации представлены транслокациями (реципрокными и робертсоновски- ми), делециями, инсерциями, инверсиями (парацентрическими и перицентрическими), кольцами и изохромосомами.
1.2 Структурные хромосомне мутации. Причины их возникновения
Структурные мутации хромосом могут возникать только в результате разрыва хромосом с последующим воссоединением, сопровождающимся нарушением исходной конфигурации хромосом. Такие мутации могут быть сбалансированными или несбалансированными [4].
Хромосомные заболевания человека и факторы их развития и наследования
... Хромосомными болезнями (хромосомными синдромами) называются комплексы множественных врожденных пороков развития, вызываемых числовыми (геномные мутации) или структурными (хромосомные аберрации) изменениями хромосом, видимыми в световой микроскоп. Хромосомные аберрации и изменения количества хромосом, как и генные мутации, ...
При сбалансированных хромосомных мутациях нет утраты или избытка генетического материала, поэтому они не имеют фенотипических проявлений, кроме тех случаев, когда в результате разрыва хромосомы в месте разрыва оказывается функционально важный ген. В то же время у носителей сбалансированных хромосомных мутаций могут образовываться несбалансированные по хромосомному набору гаметы и как следствие этого у плода, возникшего от оплодотворения такой гаметой, хромосомный набор окажется также несбалансированным. При несбалансированном хромосомном наборе у плода развиваются тяжелые клинические проявления патологии, как правило, в виде комплекса врожденных пороков развития [7].
Структурные мутации хромосом представлены делециями, микроделеционными синдромами, дупликациями, транслокациями, инсерциями, инверсиями, изо- и кольцевыми хромосомами.
1.3 Численные хромосомне мутации. Причины их возникновения
Моносомии. Отсутствие любой аутосомы является в абсолютном большинстве случаев несовместимым с нормальным развитием и приводит к ранним спонтанным абортам. Очень редкое исключение — моносомия по хромосоме 21. Моносомия может быть результатом нерасхождения хромосом, вследствие которого появляются нулисомные гаметы, или потери хромосомы во время ее движения к полюсу клетки в анафазе.
Анэуплоидия по половым хромосомам. Моносомия по половым хромосомам приводит к образованию организма с кариотипом ХО, клиническим проявлением которого служит синдром Тернера. В 80 % случаев моносомия по хромосоме X является результатом нарушения мейоза у отца (нерасхождение хромосом Х и У и как следствие — образование нулисомных гамет).
Большинство Х0-зигот погибают в раннем эмбриогенезе.
Полиплоидия. Полиплоидные клетки содержат утроенный или учетверенный гаплоидный набор хромосом. У человека триплоидия обнаруживается иногда у спонтанных абортусов, известно также несколько случаев живорождений, но больные погибали в течение 1-го месяца жизни. Триплоидия может быть обусловлена нарушением мейотического расхождения всего набора хромосом в мейозе женских (отсутствие первого мейотического деления ооцита) или мужских половых клеток. В результате либо яйцеклетка, либо сперматозоид оказываются диплоидными. В качестве механизма триплоидии рассматривают также возможность оплодотворения яйцеклетками двумя сперматозоидами. В том случае, когда триплоидия обусловлена отцовским диплоидным набором хромосом, возникает пузырное перерождение плаценты, так называемый пузырный занос [4].
Трнсомии. Трисомией называют появление в кариотипе дополнительной хромосомы. Самым известным примером трисомии является болезнь Дауна, которую часто называют трисомией по хромосоме 21. Результатом трисомии по хромосоме 13 является синдром Патау, а по хромосоме 18 — синдром Эдвардса. Все названные трисомии — аутосомные. Другие трисомики по аутосомам нежизнеспособны, погибают внутриутробно и, по-видимому, теряются в виде спонтанных абортов. Жизнеспособными являются индивидуумы с дополнительными половыми хромосомами. Более того, клинические проявления дополнительных хромосом X или У могут быть весьма незначительными.
Обычно трисомии возникают из-за нарушения расхождения гомологичных хромосом в анафазе мейоза I. В результате в одну дочернюю клетку попадают обе гомологичные хромосомы, а во вторую дочернюю клетку не попадает ни одна из хромосом бивалента (такую клетку называют нулисомной).
Метаболический синдром у детей и подростков
... органов. Кроме хронической гипертензии, патология провоцирует сахарный диабет, тяжелые поражения печени. Нередко метаболический синдром у ребенка может в последующем стать причиной ишемии сердца, которая, в свою очередь, осложняется ... Метаболический синдром у детей – это последствие влияния нескольких негативных факторов, одновременно нарушающих важные для полноценного обмена ...
Иногда, однако, трисомия может быть результатом нарушения расхождения сестринских хроматид в мейозе II. В этом случае в одну гамету попадают две совершенно одинаковые хромосомы, что в случае ее оплодотворения нормальным спермием даст трисомную зиготу. Этот тип хромосомных мутаций, ведущих к трисомии, называют нерасхождением хромосом. Отличия в исходах нарушения расхождения хромосом в мейозе I и II иллюстрирует рис. 1.1. Аутосомные трисомии возникают из-за нерасхождения хромосом, наблюдающегося преимущественно в оогенезе, но и в сперматогенезе нерасхождение аутосом также может быть. Нерасхождение хромосом может происходить и на ранних стадиях дробления оплодотворенной яйцеклетки. В этом случае в организме присутствует клон мутантных клеток, который может захватывать большую или меньшую часть органов и тканей и иногда давать клинические проявления, сходные с теми, которые наблюдают при обычной трисомии [3].
Причины нерасхождения хромосом остаются неясными. Известный факт связи между нерасхождением хромосом (особенно хромосомы 21) и возрастом матери до сих пор не имеет однозначной интерпретации. Некоторые исследователи полагают, что это может быть связано со значительным промежутком времени между конъюгацией хромосом и образованием хиазм, которые происходят у плода женского пола, т.е. достаточно рано и с расхождением хромосом в диакинезе, наблюдающемся у женщин в детородном возрасте. Следствием старения ооцитов могут быть нарушение образования веретена и другие нарушения механизмов завершения мейоза I. Рассматривается также версия об отсутствии образования хиазм в мейозе I у плодов женского пола, которые необходимы для последующего нормального расхождения хромосом [4].
ГЛАВА 2. ХАРАКТЕРИСТИКА СИНДРОМА ДАУНА
2.1 Клинико-цитогенетические характеристики синдрома Дауна
Синдром Дауна, трисомия 21 (рис. 2.1), — наиболее изученная хромосомная болезнь. Частота синдрома Дауна среди новорождённых равна 1:700—1:800, не имеет какой-либо временной, этнической или географической разницы у родителей одинакового возраста. Частота рождения детей с синдромом Дауна зависит от возраста матери и в меньшей мере от возраста отца (рис. 2.2).
С возрастом существенно возрастает вероятность рождения детей с синдромом Дауна. Так, в возрасте 45 лет она составляет около 3%. Высокая частота детей с синдромом Дауна (около 2%) наблюдается у рано рожающих женщин (до 18 лет).
Следовательно, для популяционных сравнений частоты рождения дедетей с синдромом Дауна надо принимать во внимание распределение рожающих женщин по возрасту (доля женщин, рожающих после 30—35 лет, среди всех рожающих).
Это распределение меняется иногда в течение 2—3 лет для одного и того же населения (например, при резком изменении экономической ситуации в стране).
В связи с уменьшением в 2 раза числа женщин, рожающих после 35 лет, в последние 15 лет в Белоруссии и России число детей с синдромом Дауна снизилось на 17—20%. Увеличение частоты с увеличением материнского возраста известно, но в то же время необходимо понимать, что большинство детей с синдромом Дауна рождены матерями, возраст которых младше 30 лет. Это связано с большим числом беременностей в этой возрастной группе по сравнению со старшей группой.
Посткастрационный синдром у женщин
... нарушения при П. с. требуют длительной заместительной терапии. Посткастрационный синдром у женщин репродуктивного возраста развивается главным образом после тотальной или субтотальной овариэктомии ... Это вызывает патологические симптомы, весьма сходные с симптомами при климактерическом синдроме. Нарушения секреции нейропептидов гипоталамуса (люлиберина, тиролиберина, кортиколиберина и др.) изменяют ...
В литературе описана «пучковость» рождения детей с синдромом Дауна в определённые промежутки времени в некоторых странах (городах, провинциях).
Эти случаи можно объяснить скорее стохастическими колебаниями спонтанного уровня нерасхождения хромосом, чем воздействием предполагаемых этиологических факторов (вирусная инфекция, низкие дозы радиации, хлорофос).
Цитогенетические варианты синдрома Дауна разнообразны. Однако основную долю (94—95%) составляют случаи простой полной трисомии 21 как следствие нерасхождения хромосом в мейозе. При этом материнский вклад нерасхождения в эти гаметические формы болезни составляет 80%, а отцовский — только 20%. Причины такой разницы неясны. Небольшая (около 2%) доля детей с синдромом Дауна имеет мозаичные формы D7+21/46).
Примерно 3—4% больных с синдромом Дауна имеют транслокационную форму трисомии по типу робертсоновских транслокаций между акроцентриками (D/21 и G/21).
Почти 50% транслокационных форм наследуется от родителей-носителей и 50% — транслокации, возникшие de novo.
Соотношение мальчиков и девочек среди новорождённых с синдромом Дауна составляет 1:1.
Клиническая симптоматика синдрома Дауна разнообразна: это и врождённые пороки развития, и нарушения постнатального развития нервной системы, и вторичный иммунодефицит и др. Дети с синдромом Дауна рождаются в рок, но с умеренно выраженной пренатальной гипоплазией (на 8—10% ниже средних величин).
Многие симптомы синдрома Дауна заметны при рождении, в последующем они проявляются более чётко. Квалифицированный педиатр ставит правильный диагноз синдрома Дауна в родильном доме не менее чем в 90% случаев. Из черепно-лицевых дизморфий отмечаются монголоидный разез глаз (по этой причине синдром Дауна долго называли монголоидизмом), круглое уплощённое лицо, плоская спинка носа, эпикант, крупный (обычно высунутый) язык, брахицефалия, деформированные ушные раковины (рис. 2.3) [2].
На трёх рисунках представлепредставлены фотографии детей разного возраста, и у всех имеются арактерные черты и признаки дизэмбриогенеза. Характерна мышечная гипотония в сочетании с разболтанностью суставов. Часто встречаются врождённый порок сердца, клинодактилия, характерные изменения дерматоглифики (четырёхпальцевая, или «обезьянья», складка на ладони — рис. 2.4, две кожные складки вместо трёх на мизинце, высокое положение трирадиуса и др.).
Пороки ЖКТ наблюдаются редко. Частота какого-либо симптома в 100% случаев, кроме низкого роста, не отмечена.
В табл. 2.1 и 2.2 представлена частота внешних признаков синдрома Дауна и основных врождённых пороков внутренних органов.
Диагноз синдрома Дауна ставится на основании частоты сочетания нескольких симптомов. Следующие 10 признаков наиболее важны для постановки диагноза, наличие 4—5 из которых достоверно указывает на синдром Дауна:
1) уплощение профиля лица (90%);
2) отсутствие сосательного рефлекса (85%);
3) мышечная гипотония (80%);
4) монголоидный разрез глаз (80%);
5) избыток кожи на шее (80%);
6) разболтанность суставов (80%);
7) диспластичный таз G0%);
8) диспластичные (деформированные) ушные раковины D0%);
9) клинодактилия мизинца F0%);
10) четырёхпальцевая сгибательная складка (поперечная линия) на ладони D0%).
Курсовая работа синдром дауна
... их детей. Первая группа в США, Монголоидный Совет Развития, изменила своё название на «Национальная ассоциация синдрома Дауна» в ... века причины синдрома Дауна оставались неизвестными, однако была известна взаимосвязь между вероятностью рождения ребёнка с синдромом Дауна и возрастом ... и вспомогательным медицинским персоналом, выполняют некоторую офисную работу, трудятся в кафе и супермаркетах. В Беларуси ...
Большое значение для диагностики имеет динамика физического и умственного развития ребёнка. При синдроме Дауна и то и другое задерживается. Рост взрослых больных на 20 см ниже среднего. Задержка в умственном развитии достигает имбецильности, если не применяются специальные методы обучения. Дети с синдромом Дауна ласковые, внимательные, послушные, терпеливые при обучении. Коэффициент умственного развития (IQ) у разных детей широко варьирует (от 25 до 75).
Реакция детей с синдромом Дауна на факторы окружающей среды часто патологическая в связи со слабым клеточным и гуморальным иммунитетом, снижением репарации ДНК, недостаточной выработкой пищеварительных ферментов, ограниченными компенсаторными возможностями всех систем. По этой причине дети с синдромом Дауна часто болеют пневмониями, тяжело переносят детские инфекции. У них отмечается недостаток массы тела, выражен авитаминоз [6].
Врождённые пороки внутренних органов, сниженная приспособленность детей с синдромом Дауна часто приводят к летальному исходу в первые 5 лет. Следствием изменённого иммунитета и недостаточности репарационных систем (для повреждённой ДНК) являются лейкозы, часто встречающиеся у больных с синдромом Дауна.
Дифференциальная диагностика проводится с врождённым гипотиреозом, другими формами хромосомных аномалий. Цитогенетическое исследование у детей показано и при подозрении на синдром Дауна, и при клинически установленном диагнозе, поскольку цитогенетическая характеристика пациента необходима для прогноза здоровья будущих детей у родителей и их родственников.
Этические проблемы при синдроме Дауна многоплановы. Несмотря на повышение риска рождения ребёнка с синдромом Дауна и другими хромосомными синдромами, врач должен избегать прямых рекомендаций по планированию беременности у женщин старшей возрастной группы, так как возрастной риск остаётся достаточно низким, особенно с учётом возможностей пренатальной диагностики. Неудовлетворённость у пациентов часто вызывает форма сообщения о синдроме Дауна у ребёнка. Поставить диагноз синдрома Дауна по фенотипическим признакам обычно можно немедленно после родоразрешения. Важно сообщить родителям по крайней мере о подозрениях как можно скорее после родоразрешения. Нецелесообразно полностью информировать родителей ребёнка с синдромом Дауна немедленно после родоразрешения. Нужно дать достаточно сведений, чтобы ответить на их немедленные вопросы и поддерживать их до того дня, когда станет возможно более детальное обсуждение. Немедленная информация должна включать объяснение этиологии синдрома для исключения взаимных обвинений супругов и описание исследований и процедур, необходимых для того, чтобы полностью оценить здоровье ребёнка.
Полное обсуждение диагноза нужно провести, как только родители, по крайней мере частично, оправятся от стресса родоразрешения, обычно в пределах 1-х суток. К этому времени у них возникает комплекс вопросов, на которые необходимо отвечать точно и определённо. На эту встречу приглашают обоих родителей. В этот период ещё слишком рано нагружать родителей всей информацией о заболевании, так как эти новые и сложные понятия требуют времени для восприятия.
Лечебная помощь детям с синдромом Дауна многопланова и неспецифична. Врождённые пороки сердца устраняют оперативно. Постоянно проводится общеукрепляющее лечение. Питание должно быть полноценным. Необходимы внимательный уход за больным ребёнком, защита от действия вредных факторов окружающей среды (простуда, инфекции).
Эдвардс Синдромы Қазақша
... без . . . (даун, эдвардс, Патау, Клайнфельтер, Тернер синдромдары) дамиды . Дәріскер . . Даун синдромы — Уикипедия, қазақша . Айтыс қазақша реферат скачать бесплатно . Все эти реакции . . Характерное для синдрома Эдвардса положение пальцев (возраст ребенка 2 месяца) . Аса ...
Многие больные с трисомией 21 теперь способны вести самостоятельную жизнь, овладевают несложными профессиями, создают семи [2].
2.2 Пример развёрнутой клинической характеристики синдрома Дауна
Период новорождённости
Немедленно после родоразрешения ребёнка полностью обследуют, чтобы подтвердить диагноз и выявить все неотложные медицинские проблемы. В большинстве ситуаций необходима соответствующая консультация педиатра.
Сердечно-сосудистая система
Врождённый порок сердца, обычно в форме эндокардиальных дефектов, отмечается у 40% новорождённых и должен быть исключен эхокардиографическим скринингом вскоре после рождения, поскольку такие пороки трудно обнаружить. Встречаются также септальные дефекты и тетрада Фалло. Обнаружение серьёзных врождённых пороков развития часто диктует необходимость хирургического вмешательства. Важно подчеркнуть, что ребёнку с синдромом Дауна требуется точно такое же медицинское и хирургическое лечение, как и ребёнку без хромосомного нарушения. Серьёзный врождённый порок сердца остаётся главной причиной смерти детей с синдромом Дауна, несмотря на прогресс в хирургическом лечении.
При отсутствии врождённого сердечного дефекта большинство пациентов могут дожить до 6-го десятилетия.
ЖКТ
Наиболее частое врождённое расстройство ЖКТ, сочетающееся с синдро-мом Дауна, — атрезия двенадцатиперстной кишки, хотя описаны и пилорический стеноз, болезнь Хиршспрунга и трахеоглоточный свиш. Хирургическое вмешательство и в этом случае должно быть произведено независимо от хромосомного нарушения. Общая частота пороков развития ЖКТ — приблизительно 12%.
Зрение
У 3% новорождённых с синдромом Дауна имеются плотные врождённые катаракты, которые должны быть рано удалены. Также более часто встречается глаукома.
Вскармливание
Гипотония — постоянный симптом у новорождённых с синдромом Дауна. Эта слабость может мешать грудному вскармливанию, и, возможно, должен быть привлечен опытный консультант по лактации, чтобы гарантировать, что процесс успешен. Вскармливание имеет тенденцию занимать больше времени, и могут быть проблемы приложения к груди из-за выступающего языка. У некоторых новорождённых не поддерживается необходимая температура тела и они могут нуждаться в дополнительном пеленании во время кормления. Более часты запоры из-за гипотонической мускулатуры кишечника.
Врождённый гипотиреоз
Это заболевание несколько более распространено среди новорождённых с синдромом Дауна. Его обнаруживают при массовом скрининге, выполняемом всем новорождённым.
Врождённый вывих бедра
Обшая мышечная слабость и гипотония увеличивают частоту вывиха бедра, хотя истинный врождённый вывих весьма редок. На это необходимо обратить дополнительное внимание в ходе обычного обследования новорождённого [1,2,5].
Грудной возраст
Как только проведены все неотложные медицинские мероприятия и успешно начато вскармливание, родители могут взять новорождённого домой. Если участковый (семейный) врач не привлекался во время пребывания больного в стационаре, ему необходимо рано войти в контакт с семьёй, важно провести оценку начального медицинского состояния ребёнка. Этот «хороший педиатрический осмотр» означает, что врач не должен оказаться впервые перед незнакомым и очевидно больным ребёнком несколькими месяцами позже.
Медицинское обслуживание на 1-м году жизни включает постоянное на-блюдение с учётом проблем, выявленных в периоде новорождённости, а также обследования для выявления приобретённых проблем, типа нарушений слуха или зрения. Ранний и регулярный контакт с соответствующими опытными консультантами должен начаться уже на 1-м году жизни. Судорожные приступы более часты у детей с синдромом Дауна (приблизительно 10%) и могут возникать с раннего возраста. По характеру они обычно тонические или клонические. Люди с синдромом Дауна имеют сниженный клеточный иммунитет, поэтому дети, вероятно, перенесут больше инфекционных болезней дыхательных путей. Нарушения проходимости верхних дыхательных путей также более часты из-за гипертрофии миндалин и аденоидов. С изменениями в иммунитете также связано увеличение частоты лейкозов у людей с синдромом Дауна, хотя эта связь неясна. В практическом смысле снижение иммунитета имеет небольшое значение. В обычное время должна быть начата нормальная профамма прививок. Теперь повсеместно принята практика «раннего вмешательства» как имеющая выгоды для ребёнка и семьи. Это означает домашнее или стационарное лечение поражённого ребёнка с очень раннего возраста с участием врачей-специалистов, физиотерапевтов и логопедов. Родители также должны участвовать в лечении ребёнка. Родители должны понимать, что официальная программа помощи предпочтительнее так называемых маргинальных врачей, которые могут истощать родительские ресурсы без видимых результатов [1,2,5].
Детство
По мере роста ребёнка в дошкольном периоде становится очевидным, что развитие в целом запаздывает. Физические параметры будут отставать от нормы из-за гипотонии и общей слабости, речь, вероятно, будет затруднена и может быть поздняя социальная адаптация. Психометрическая оценка показывает, что у большинства детей с синдромом Дауна интеллектуальное функционирование повреждено умеренно, но возможный диапазон интеллектуального дефекта огромен. В это время полезно помочь родителям в осознании того, что для этого ребёнка являются уместными другие параметры и что сравнение развития ребёнка с сибсами (братьями и сестрами) не окажет большой помощи. Сравнение с другими родителями детей с синдромом Дауна полезно, но нужно помнить, что каждый ребёнок развивается по-своему. Важно не делать слишком много предсказаний относительно того, насколько хорошо и быстро ребёнок разовьется, и строить своё поведение на основе сдержанного оптимизма и разумных ожиданий, как это касается всех детей. К этому времени должны хорошо развиться пожизненные отношения практикующего врача с ребёнком. Хорошее знание врачом того, что является нормальным для такого ребёнка, позволит рано распознать любые проблемы со здоровьем, особенно в отношении более частых при этом состоянии болезней. Кроме того, однако, проницательный врач будет помнить, что у ребёнка с синдромом Дауна могут возникнуть те же проблемы, что и у любого другого, и что не все симптомы будут вызваны этим синдромом.
Подход практикующего врача к ребёнку с синдромом Дауна должен быть точно таким же, как к любому другому ребёнку: дружественный, неугрожаюший и диалоговый. Родители — обычно неоценимые источники информации относительно ребёнка и после нескольких лет тяжёлой и кропотливой работы станут верными защитниками и «борцами с бюрократией». Их беспокойства должны быть рассмотрены с должным вниманием.
Будучи вовлечёнными в ранние дошкольные программы, большинство детей с синдромом Дауна хорошо подготовлены для поступления в основное обучение в обычное время. Задача врача — поддержать родителей в принятии их решения и устранить любые медицинские проблемы, которые могут возникать при выборе школы.
Врождённый порок сердца
Серьёзные пороки развития, которые не могут быть полностью вылечены, остаются главной причиной смерти в детстве. Необходимо поддерживать тесную связь с педиатром-кардиологом.
Сенсорный дефицит
Существенные ухудшения слуха встречаются у большинства детей с синдромом Дауна. Рекомендуются ежегодная аудиометрия и консультация специалиста.
Также часто встречается ухудшение зрения из-за нарушений рефракции или косоглазия, и детей должен ежегодно осматривать офтальмолог. Часто развиваются катаракты, но обычно вне визуальной оси.
Гипотиреоз
В связи с высокой частотой (до 30%) гипотиреоз должен быть исключен на основе стандартной скрининговой процедуры. Хотя в большинстве случаев заболевание развивается в подростковом периоде, детям более ранних возрастов рекомендуется биохимической скрининг 1 раз в 2 года. Если обнаружены любые признаки болезни щитовидной железы, необходимо раннее обследование и лечение.
Атлантоаксиальная неустойчивость
До 15% детей с синдромом Дауна имеет рентгенологическое подтверждение неустойчивости атлантоаксиального соединения, но только в небольшой части случаев это приводит к сдавлению спинного мозга с неврологической симптоматикой. Возникает вопрос: необходимо ли массовое рентгенологическое обследование всем людям с синдромом Дауна и если да, то в каком возрасте? Когда неустойчивость обнаружена, служит ли это показанием для ограничения спортивных занятий и местных воздействий в попытке предотвратить редкое осложнение повреждения спинного мозга? У людей с синдромом Дауна иногда трудно обнаружить тонкие неврологические симптомы и может потребоваться хирургическое вмешательство для стабилизации этой области.
На настоящий момент мнения сходятся в пользу рентгенологического скрининга перед поступлением в школу, главным образом чтобы убедить родителей подавляющего большинства детей в отсутствии у последних неустойчивости атлантоаксиального соединения. Если найдены неустойчивость или анатомические расстройства, то рекомендации должны быть достаточно осторожными, чтобы гарантировать, что потенциальные воздействия соответственно изменены, но без излишнего ограничения ребёнка. Необходимо неврологическое наблюдение.
Физическое развитие
Физическое развитие у детей с синдромом Дауна неизменно задержано, и для точного контроля необходимо использовать модифицированные процентильные диаграммы роста и массы тела. Тенденция к ожирению требует специального внимания в этой группе детей к здоровой диете и занятиям лечебной физкультурой.
Стоматологическая профилактика
У детей с синдромом Дауна часто маленькие и деформированные зубы. Чтобы гарантировать адекватный зубной ряд для взрослой жизни, требуются ранние и частые осмотры зубов начиная со 2-го года жизни [1,2,5].
Пубертатный период
У ребенка с синдромом Дауна также наступает гормональная перестройка, которая обычно сопровождает пубертатный период. Все обычные трудности и проблемы этой потенциально трудной стадии развития должна быть рассмотрены. Это включает проблемы подростка, пытающегося установить свою собственную идентичность, найти своё место в жизни и преследующего свои собственные интересы.
Инвалиды — половые существа, и лица с синдромом Дауна не являются исключением. Серьёзная ошибка — поддержание стереотипа рассматривать людей с синдромом Дауна как «счастливых вечных детей». Подростки с синдромом Дауна подчинены тем же самым желаниям и эмоциям. как остальные, хотя они часто больше страдают в их выражении. Определённые медицинские состояния нуждаются во внимании врача.
Менструация и различие полов (сексуальность)
Менархе у девушек с синдромом Дауна обычно только слегка задержано. Менструации обычно устанавливаются регулярные, и, хотя большинство циклов будут ановуляторными, должна предполагаться беременность. В мировой литературе имеются описания около 30 случаев беременности у женшин с синдромом Дауна. Имеется множество описаний женщин с синдромом Дауна, менструации и плодовитость которых регулировались с помощью медикаментозной терапии типа прогестерона или хирургического вмешательства.
Гипотиреоз
Поскольку большинство случаев гипотиреоза при синдроме Дауна развивается в подростковом периоде, необходимы ежегодные исследования функции щитовидной железы.
Кожа
Кожа детей с синдромом Дауна имеет тенденцию к сухости и экземе. В течение пубертатного периода часто появляются фолликулит и угревая сыпь [1,2,5].
ГЛАВА 3. ЦИТОГЕНЕТИЧЕСКАЯ ХАРАКТЕРИСТИКА И КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ СИНДРОМА ПАТАУ
хромосомный мутация синдром даун
Синдром Патау выделен в самостоятельную нозологическую форму в 1960 г. в результате генетического исследования, проведённого у детей с врождёнными пороками развития. Частота синдрома Патау среди новорождённых равна 1:5000—1:7000. Цитогенетические варианты этого синдрома следующие. Простая полная трисомия 13 (рис. 3.1) как следствие нерасхождения хромосом в мейозе у одного из родителей (главным образом у матери) встречается у 80—85% больных. Остальные случаи обусловлены в основном передачей дополнительной хромосомы (точнее, её длинного плеча) в робертсоновских транслокациях типа D/13 и G/13. Обнаружены и другие цитогенетические варианты (мозаицизм, изохромосома, неробертсоновские транслокации), но они встречаются крайне редко. Клиническая и патологоанатомическая картина простых трисомных форм и транслокационных не различается.
Соотношение полов при синдроме Патау близко к 1:1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25—30% ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок гестации 38,3 нед).
Характерное осложнение беременности при вынашивании плода с синдромом Патау — многоводие: оно встречается почти в 50% случаев синдрома Патау.
Для синдрома Патау характерны множественные врождённые пороки развития головного мозга и лица (рис. 3.2).
Это патогенетически единая группа ранних (и, следовательно, тяжёлых) нарушений формирования головного мозга, глазных яблок, мозговой и лицевой частей черепа. Окружность черепа обачно уменьшена, встречается и тригоноцефалия. Лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположенные и деформированные. Типичный признак синдрома Патау — расщелины верхней губы и нёба (обычно двусторонние).
Всегда обнаруживаются пороки некольких внутренних органов в разной комбинации: дефекты перегородок сердца, незавершённый поворот кишечника, кисты почек, аномалии внутренних половых органов, дефекты поджелудочной железы. Как правило, наблюдаются полидактилия (чаще двусторонняя и на руках) и флексорное положение кистей. Частота разных симптомов у детей с синдромом Патау представлена в табл. 3.1.
Клиническая диагностика синдрома Патау основывается на сочетании характерных пороков развития. При подозрении на синдром Патау показано УЗИ всех внутренних органов.
В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95% — до I года).
Однако некоторые больные живут несколько лет. Более того, в развитых странах отмечается тенденция увеличения продолжительности жизни больных с синдромом Патау до 5лет (около 15% детей) и даже до Шлет B—3% детей).
Другие синдромы врождённых пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по отдельным признакам совпадают с синдромом Патау. Решающий фактор в диагностике — исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей в семье.
Лечебная помощь детям с синдромом Патау неспецифическая: операции по поводу врождённых пороков развития (по жизненным показаниям), общеукрепляющее лечение, тщательный уход, профилактика простудных и инфекционных болезней. Дети с синдромом Патау практически всегда имеют глубокую идиотию [2,5].
ГЛАВА 4. ЦИТОГЕНЕТИЧЕСКАЯ ХАРАКТЕРИСТИКА И КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ СИНДРОМА ЭДВАРДСА
Это трисомии по 18 хромосоме (рис. 4.1).
Почти во всех случаях синдром Эдвардса обусловлен простой трисомной формой (гаметическая мутация у одного из родителей).
Встречаются и мозаичные формы (нерасхождение на ранних стадиях дробления).
Транслокационные формы крайне редки и, как правило, это частичные, а не полные трисомии. Клинических различий между цитогенетически различающимися формами трисомии нет. Частота синдрома Эдвардса составляет 1:5000—1:7000 новорождённых. Соотношение мальчиков и девочек равно 1:3. Причины преобладания больных девочек пока неясны. При синдроме Эдвардса отмечается выраженная задержка пренатального развития при полной продолжительности беременности (роды в срок).
На рис. 4.2 представлены пороки развития, характерные для синдрома Эдвардса. В первую очередь это множественные врождённые пороки развития лицевой части черепа, сердца, костной системы, половых органов. Череп долихоцефалической формы; нижняя челюсть и отверстие рта маленькие; глазные щели узкие и короткие; ушные раковины деформированные и низко расположенные. Из других внешних признаков отмечаются флексорное положение кистей, аномально развитая стопа (пятка выступает, свод провисает), I палец стоп короче II. Спинномозговая грыжа и расщелина губы встречаются редко E% случаев синдрома Эдвардса).
Многообразная симптоматика синдрома Эдвардса у каждого больного проявляется лишь частично. Частота отдельных врождённых пороков приведена в табл. 4.1.
Как видно из табл. 5.5, наиболее значимыми в диагностике синдрома Эдвардса являются изменения мозгового черепа и лица, опорно-двигательного аппарата, пороки развития сердечно-сосудистой системы. Дети с синдромом Эдвардса умирают в раннем возрасте (90% — до 1 года) от осложнений, обусловленных врождёнными пороками развития (асфиксия, пневмония, кишечная непроходимость, сердечно-сосудистая недостаточность).
Клиническая и даже патологоанатомическая дифференциальная диагностика синдрома Эдвардса сложна. Во всех случаях показано цитогенетическое исследование [2,5].
ВЫВОДЫ
- В данной работе был проведен анализ синдромов, вызванных трисомией аутосом: трисомии X)">синдром Дауна – трисомии 21, синдром Патау – трисомии 13, синдром Эдвардса – трисомии 18, их цитогенетическая и клиническая характеристика.
- Трисомией называют появление в кариотипе дополнительной хромосомы. Самым известным примером трисомии является болезнь Дауна, которую часто называют трисомией по хромосоме 21.
- Обычно трисомии возникают из-за нарушения расхождения гомологичных хромосом в анафазе мейоза I. Иногда, однако, трисомия может быть результатом нарушения расхождения сестринских хроматид в мейозе II.
- Синдром Дауна, трисомия 2 – наиболее изученная хромосомная болезнь. Частота синдрома Дауна среди новорождённых равна 1:700—1:800.
— Цитогенетические варианты синдрома Дауна разнообразны. Однако основную долю (94—95%) составляют случаи простой полной трисомии 21 как следствие нерасхождения хромосом в мейозе. Соотношение мальчиков и девочек среди новорождённых с синдромом Дауна составляет 1:1.
— Синдром Патау выделен в самостоятельную нозологическую форму в 1960 г. в результате генетического исследования, проведённого у детей с врождёнными пороками развития. Частота синдрома Патау среди новорождённых равна 1:5000—1:7000. Цитогенетические варианты этого синдрома следующие. Простая полная трисомия 13 как следствие нерасхождения хромосом в мейозе у одного из родителей (главным образом у матери) встречается у 80—85% больных. Соотношение полов при синдроме Патау близко к 1:1.
- Синдром Эдвардса – это трисомии по хромосоме номер 18. Почти во всех случаях синдром Эдвардса обусловлен простой трисомной формой.
Встречаются и мозаичные формы.Частота синдрома Эдвардса составляет 1:5000—1:7000 новорождённых. Соотношение мальчиков и девочек равно 1:3.
- Многообразная симптоматика синдрома Эдвардса у каждого больного проявляется лишь частично.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
[Электронный ресурс]//URL: https://psychoexpert.ru/kursovaya/na-temu-sindromyi/
1. Бердышев Г.Д., Криворучко И.Ф. Генетика человека. – К.: Вища школа, 1979 – 447с.
2. Бочков Н. П. Клиническая генетика: Учебник. – 2-е изд. перераб. и доп. – М.: ГЕОТАР-МЕД, 2002 – 448с.
3. Генетика. Учебник для вузов/ Под ред. Скадемика РАМН В. И. Иванова. – М.: ИКЦ «Академкнига», 2006. – 638с.
4. Гинтер Е. К. Медицинская генетика: Учебник. – М.: Медицина, 2003 – 448с.
5. Гречаніна О.Я. Медична генетика. – К.: Медицина, 2007
6. Запорожан В.М. Медична генетика. – Одеса: Одеський мед університет. – 2005
7. Фогель Ф., Мотульски А. Генетика человека: В 3-х Т.: Пер. с англ. – М.: Мир, 1989
ПРИЛОЖЕНИЕ
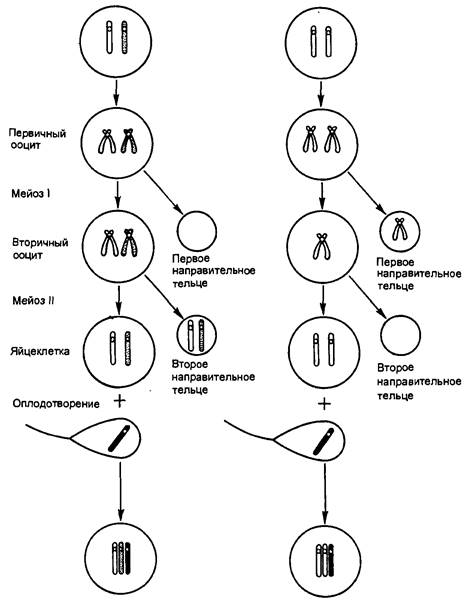
Нерасхождение в мейозе IНерасхождение в мейозе II
Рис. 1.1 – Мейотическое нерасхождение.
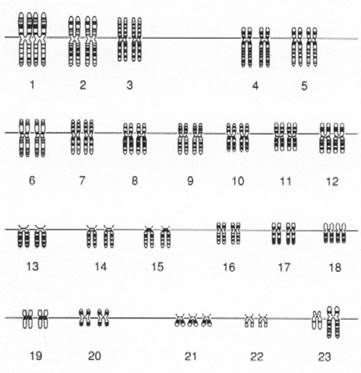
Рис. 2.1 – трисомия по хромосоме 21.
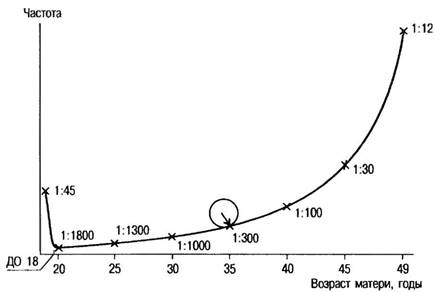
Рис. 2.2 – Зависимость частоты рождения детей с синдромом Дауна от возраста матери.

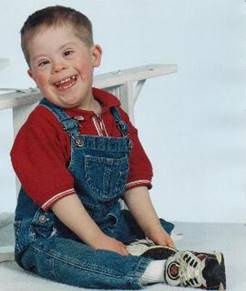
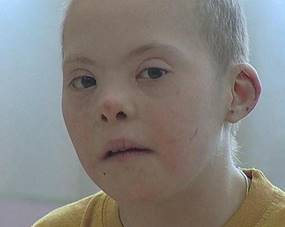

Рис. 2.3 – Дети разного возраста с характерными чертами синдрома Дауна.
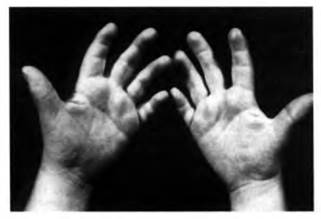
Рис. 2.4 – Ладони взрослого мужчины с синдромом Дауна.
Таблица2.1 — Наиболее частые внешние признаки синдрома Дауна (по Г. И. Лазюку с доп.)
| Порок или признак | Частота, % общего числа больных |
| Мозговой череп и лицо | 98,3 |
| Брахицефалия | 81,1 |
| Монголоидный разрез глазных щелей | 79,8 |
| Эпикант | 51,4 |
| Плоская спинка носа | 65,9 |
| Узкое нёбо | 58,8 |
| Большой высунутый язык | 9 |
| Деформированные ушные раковины | 43,2 |
| Костно-мышечная система, конечности | 100,0 |
| Низкий рост | 100,0 |
| Деформация грудной клетки | 26,9 |
| Короткие и широкие кисти | 64,4 |
| Клинодактилия мизинца | 56,3 |
| Укороченная средняя фаланга V пальца кисти с одной сгибательной складкой | ? |
| Четырёхпальцевая складка на ладони | 40,0 |
| Сандалевидная щель | ? |
| Глаза | 72,1 |
| Пятна Брашфилда | 68,4 |
| Помутнение хрусталика | 32,2 |
| Косоглазие | 9 |
Таблица 2.2 — Основные врождённые пороки внутренних органов при синдроме Дауна (по Г. И. Лазюку с дополнениями)
| Пораженная система и порок | Частота % общего числа больных |
| Сердечно-сосудистая система | 53,2 |
| Дефект межжелудочковой перегородки | 31,4 |
| Дефект межпредсердной перегородки | 24,3 |
| Открытый атриовентрикулярный канал | 9 |
| Аномалии крупных сосудов | 23,1 |
| Органы пищеварения | 15,3 |
| Атрезия или стеноз двенадцатиперстной кишки | 6,6 |
| Атрезия пищевода | 0,9 |
| Атрезия прямой кишки и ануса | 1,1 |
| Мегаколон | 1,1 |
| Мочевая система (гипоплазия почек, гидроуретер, гидронефроз) | 5,9 |
Рис. 3.1 – трисомия по 13 хромосоме
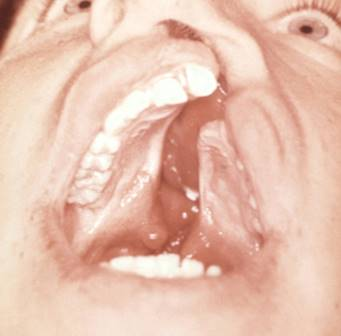
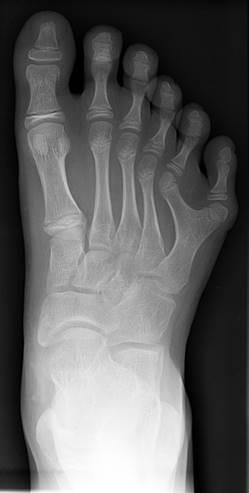
Рис. 3.2 – характерные признаки больных синдромом Патау.
Таблица3.1 — Основные врождённые пороки при синдроме Патау (по Г. И. Лазюку)
| Пораженная система и порок | Относительная частота, % |
| Лицо и мозговой череп | 96,5 |
| низко расположенные и(или) деформированные ушные раковины | 80,7 |
| расщелина верхней губы и нёба | 68,7 |
| в том числе только нёба | 10,0 |
| микрогения | 32,8 |
| дефект скальпа | 30,8 |
| Опорно-двигательный аппарат | 92,6 |
| полидактилия кистей | 49,0 |
| полидактилия стоп | 35,7 |
| флексорное положение кистей | 44,4 |
| стопа-качалка | 30,3 |
| ЦНС | 83,3 |
| аринэнцефалия | 63,4 |
| в том числе голопрозэнцефалия | 14,5 |
| микроцефалия | 58,7 |
| аплазия и гипоплазия мозолистого тела | 19,3 |
| гипоплазия мозжечка | 18,6 |
| в том числе гипоплазия и аплазия червя | 11,7 |
| аплазия и гипоплазия зрительных нервов и трактов | 17,2 |
| Глазное яблоко | 77,1 |
| микрофтальмия | 70,5 |
| колобома радужки | 35,3 |
| помутнение хрусталика | 25,9 |
| анофтальмия | 7,5 |
| Сердечно-сосудистая система | 79,4 |
| дефект межжелудочковой перегородки | 49,3 |
| в том числе компонент комбинированного порока | 44,8 |
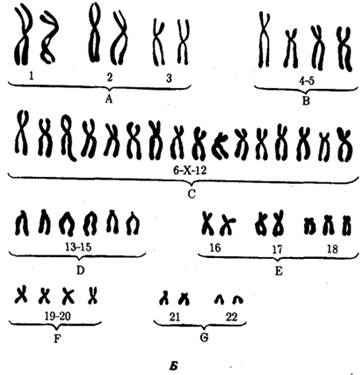
Рис. 4.1 – трисомия по 18 хромосоме.
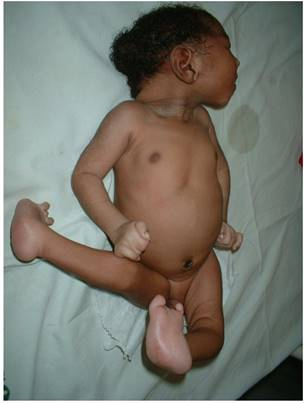
Рис. 4.2 – ребёнок с синдромом Эдвардса.
Таблица 4.1 — Основные врождённые пороки при синдроме Эдвардса (по Г. И. Лазюку)
| Пораженная система и порок (признак) | Относительная частота, % |
| Мозговой череп и лицо | 100,0 |
| микрогения | 96,6 |
| низко расположенные и(или) деформированные ушные раковины | 95,6 |
| долихоцефалия | 89,8 |
| высокое нёбо | 78,1 |
| расщелина нёба | 15,5 |
| микростомия | 71,3 |
| Опорно-двигательный аппарат | 98,1 |
| флексорное положение кистей | 91,4 |
| дистальное расположение I пальца кисти | 28,6 |
| гипоплазия и аплазия I пальца кисти | 13,6 |
| короткий и широкий I палец стопы | 79,6 |
| стопа-качалка | 76,2 |
| кожная синдактилия стоп | 49,5 |
| косолапость | 34,9 |
| короткая грудина | 76,2 |
| ЦНС | 20,4 |
| гипоплазия и аплазия мозолистого тела | 8,2 |
| гипоплазия мозжечка | 6,8 |
| Глаза (микрофтальмия) | 13,6 |
| Сердечно-сосудистая система | 90,8 |
| дефекты межжелудочковой перегородки | 77,2 |
| в том числе входящие в комбинированные пороки | 65,4 |
| дефекты межпредсердной перегородки | 25,2 |
| в том числе входящие в комбинированные пороки | 23,8 |
| аплазия одной створки клапана лёгочной артерии | 18,4 |
| аплазия одной створки клапана аорты | 15,5 |
| Органы пищеварения | 54,9 |
| дивертикул Меккеля | 30,6 |
| незавершённый поворот кишечника | 16,5 |
| атрезия пищевода | 9,7 |
| атрезия желчного пузыря и жёлчных ходов | 6,8 |
| эктопия ткани поджелудочной железы | 6.8 |
| Мочевая система | 56.9 |
| сращение почек | 27,2 |
| удвоение почек и мочеточника | 14.6 |
| кисты почек | 12,6 |
| гидро- и мегалоуретер | 9,7 |
| Половые органы | 43,5 |
| крипторхизм | 28,6 |
| гипоспадия | 9,7 |
| гипертрофия клитора | 16,6 |